Kicked off in the late 20th Century, the widely publicized Human Genome Project (HGP) set out to read our complete DNA sequence for the first time, and to define all of the genes within our genome. While the work has become foundational to biology, producing critical data and spawning countless therapeutics, its conclusion was surprising, finding only 20,000 coding sequences for proteins from the 21 million predicted open reading frames (ORFs). It seemed that only about 2% of human DNA had the potential to code for proteins, the fundamental building blocks and machines of life.
The rest of our DNA was dubbed non-coding, as it did not code for a protein as defined by the HGP. The project developed conservative rules from observations of many known proteins and statistical constraints to reduce false positives in identifying genes. The researchers defined a gene (sequences that give rise to proteins) as sequences of amino acids that are homologous to known proteins in other species and humans, are greater than 100 amino acids long, and are book-ended by standard translational start (AUG) and stop codons (UAA, UAG, UGA). Polypeptides that adhere to these rules, they concluded, can be reliably defined as proteins. However, as is often true in biology, and of rules in general, there are exceptions. Several well-understood and therapeutically important proteins, including insulin and chemokines, do not follow these rules, providing evidence that these constraints are obscuring the complete human proteome.
Although only 2% of the genome was originally annotated as protein-coding, it turns out that much of the remaining genomic DNA gives rise to RNA, the intermediary between DNA and proteins, but the function of these transcripts have since been proposed as regulatory or simply junk. The field has now established that upward of 80% of human genomic DNA is transcribed into RNA that does not arise from the original annotated genes, including classes such as long non-coding RNA (greater than 200 nucleotides long).
Over the last few decades, next-generation sequencing (NGS) and the advent of standardized tools have enabled highly parallelized, reproducible and precise detection of nucleotide sequences in the human genome and RNAs in a cell. Standard proteomics methods, on the other hand, are extremely limited in their ability to detect proteins; a large percentage of the acquired data is often discarded because the protein sequence cannot be resolved or mapped back to an annotated proteome. Due to the reliability of NGS-based approaches, RNA sequencing is often used as a surrogate for detecting protein expression. However, every RNA in a cell is not translated into a protein, which yields an inaccurate snapshot of cellular and biological function.
Introducing translatomic discovery
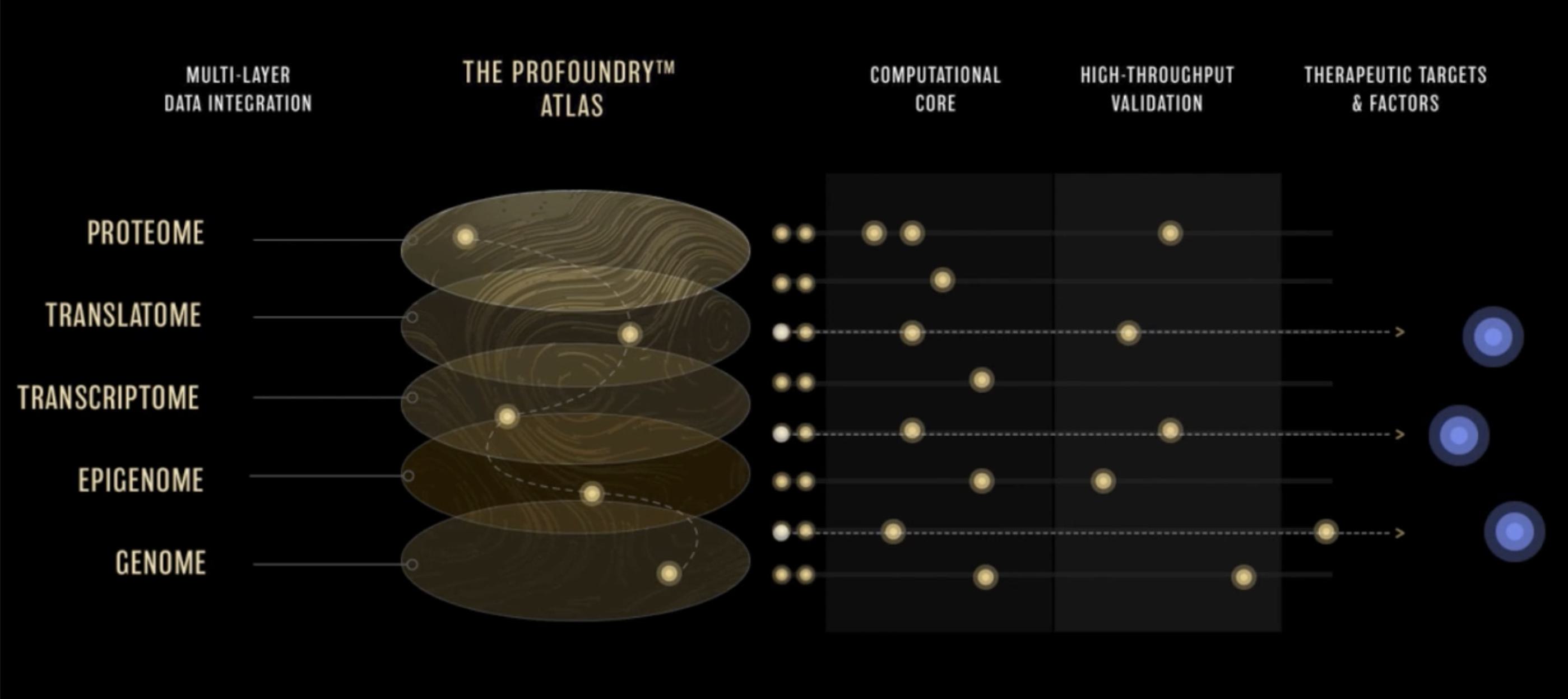
Rather than rely on imprecise detection of resulting proteins to define regions of the genome that are expressed, translatomics focuses on RNA fragments that are bound to and protected by ribosomes, the protein factories of the cell, presenting a window into the transcripts as they are actively being translated to proteins. Early ribosome profiling studies could determine if a ribosome was bound to a transcript, suggesting that it had the potential to be translated into a protein. As the technology advanced, it provided better resolution as to where on the transcript the ribosome was binding, enabling precise discovery of new ORFs that give rise to proteins, and a more unbiased view of the complete proteome. Considering the fundamental roles proteins play in biology, including structure, signalling, chemical reactions, and more, a broader set of human proteins most certainly harbors additional interesting building blocks, functional elements, and factors that we can harness to treat disease.
To tackle this challenge, Flagship-founded ProFound™ Therapeutics is taking a systematic approach to protein discovery. The company has developed the ProFoundry™ platform, which uses a combination of state-of-the-art detection, advanced computation, and high-throughput functional genomics technologies to systematically identify proteins and predict and validate their function.